Overview
Tools for analysis of RT-qPCR gene expression data using and methods, including t-tests, ANOVA, ANCOVA, repeated-measures models, and publication-ready visualizations. The package implements a general calculation method described by Ganger et al. (2017) and Taylor et al. (2019), covering both the Livak and Pfaffl methods. See the calculation method for details.
Functions
The rtpcr package gets efficiency (E) the Ct values of
genes and performs different analyses using the following functions.
| Function | Description |
|---|---|
ANOVA_DCt |
ANOVA analysis |
ANOVA_DDCt |
ANOVA analysis |
REPEATED_DDCt |
ANOVA analysis for repeated-measures data |
TTEST_DDCt |
method t-test analysis |
plotFactor |
Bar plot of gene expression for one-, two- or three-factor experiments |
Means_DDCt |
Pairwise comparison of RE values for any user-specified effect |
efficiency |
Amplification efficiency statistics and standard curves |
meanTech |
Calculate mean of technical replicates |
multiplot |
Combine multiple ggplot objects into a single layout |
Quick start
Installing and loading
The rtpcr package can be installedb by running the
following code in R:
from CRAN:
# Installing from CRAN
install.packages("rtpcr")
# Loading the package
library(rtpcr)Or from from GitHub (developing version):
devtools::install_github("mirzaghaderi/rtpcr", build_vignettes = FALSE)Input data structure
For relative expression analysis (using TTEST_DDCt,
ANOVA_DCt, ANOVA_DDCt and
REPEATED_DDCt functions), the amplification efficiency
(E) and Ct or Cq values (the mean
of technical replicates) is used for the input table. if the
E values are not available you should use ‘2’ instead
representing the complete primer amplification efficiency. The required
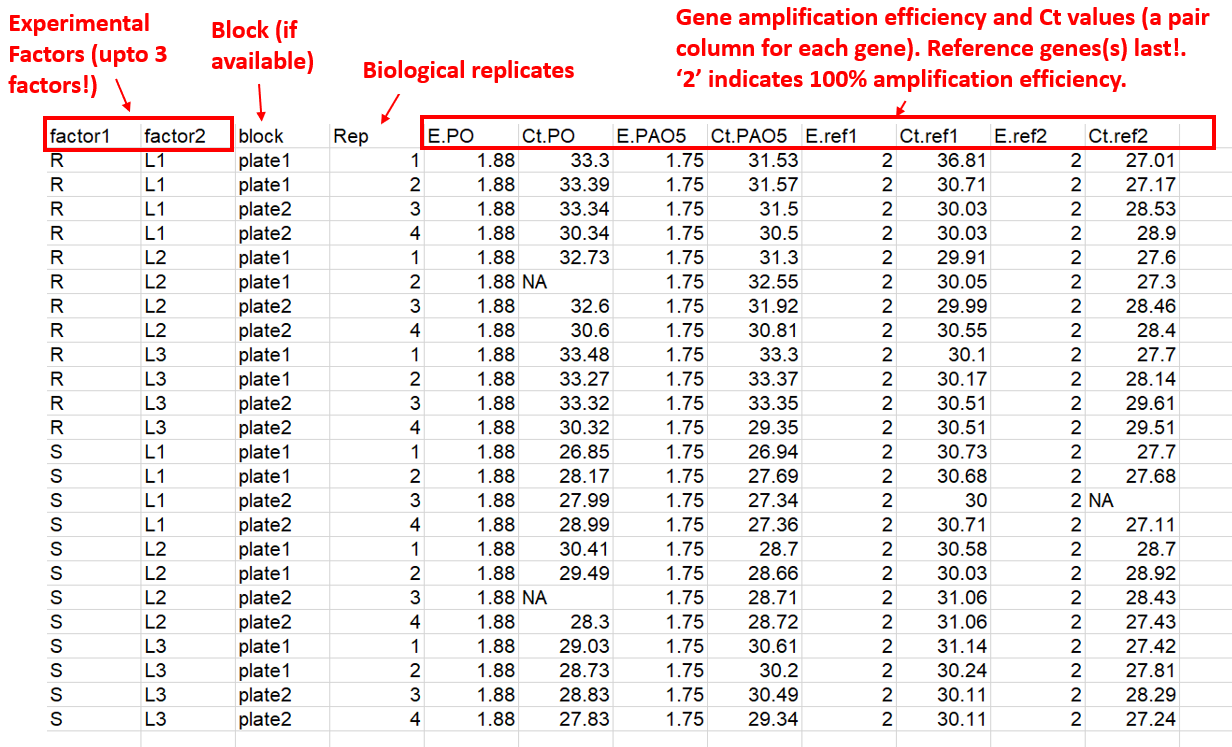
column structure of the input data is:
- Experimental condition columns (up to 3 factors, and one block if available)
- Replicates information (biological replicates or subjects; see NOTE 1, and NOTE 2)
- Target genes efficiency and Ct values (a pair column for each gene).
- Reference genes efficiency and Ct values (a pair column for each gene)
The package supports one or more target or reference gene(s), supplied as efficiency–Ct column pairs. Reference gene columns must always appear last. A sample input data is presented below.
NOTE 1
For TTEST_DDCt, ANOVA_DCt, and
ANOVA_DDCt, each row is from a separate and uniq biological
replicate. For example, a dataframe with 12 rows has come from an
experiment with 12 individuals. The REPEATED_DDCt function
is intended for experiments with repeated observations (e.g. time-course
data). For REPEATED_DDCt, the Replicate column contains
identifiers for each individual (id or subject). For example, all rows
with a 1 at Rep column correspond to a single individual,
all rows with a 2 correspond to another individual, and so
on, which have been sampled at specific time points.
NOTE 2
Your data table may also include technical replicates. In this case,
the meanTech function should be applied first to calculate
the mean of the technical replicates. The resulting table is then used
as the input for expression analysis. To use the meanTech
function correctly, the technical replicate column must appear
immediately after the biological replicate column (see Mean of technical replicates
for an example).
Data Analysis
Amplification Efficiency
The efficiency function calculates the amplification
efficiency (E), slope, and R² statistics for genes, and performs
pairwise comparisons of slopes. It takes a data frame in which the first
column contains the dilution ratios, followed by the Ct value columns
for each gene.
# Applying the efficiency function
data <- read.csv(system.file("extdata", "data_efficiency.csv", package = "rtpcr"))
data
dilutions Gene1 Gene2 Gene3
1.00 25.58 24.25 22.61
1.00 25.54 24.13 22.68
1.00 25.50 24.04 22.63
0.50 26.71 25.56 23.67
0.50 26.73 25.43 23.65
0.50 26.87 26.01 23.70
0.20 28.17 27.37 25.11
0.20 28.07 26.94 25.12
0.20 28.11 27.14 25.11
0.10 29.20 28.05 26.17
0.10 29.49 28.89 26.15
0.10 29.07 28.32 26.15
0.05 30.17 29.50 27.12
0.05 30.14 29.93 27.14
0.05 30.12 29.71 27.16
0.02 31.35 30.69 28.52
0.02 31.35 30.54 28.57
0.02 31.35 30.04 28.53
0.01 32.55 31.12 29.49
0.01 32.45 31.29 29.48
0.01 32.28 31.15 29.26
# Analysis
efficiency(data)
$Efficiency
Gene Slope R2 E
1 Gene1 -3.388094 0.9965504 1.973110
2 Gene2 -3.528125 0.9713914 1.920599
3 Gene3 -3.414551 0.9990278 1.962747
$Slope_compare
$contrasts
contrast estimate SE df t.ratio p.value
C2H2.26 - C2H2.01 0.1400 0.121 57 1.157 0.4837
C2H2.26 - GAPDH 0.0265 0.121 57 0.219 0.9740
C2H2.01 - GAPDH -0.1136 0.121 57 -0.938 0.6186Relative expression
Relative expression analysis can be done using or methods. Below is an example of expression analysis using method.
# An example of a properly arranged dataset from a repeated-measures experiment.
data <- read.csv(system.file("extdata", "data_repeated_measure_1.csv", package = "rtpcr"))
data
time id E_Target Ct_target E_Ref Ct_Ref
1 1 2 18.92 2 32.77
1 2 2 15.82 2 32.45
1 3 2 19.84 2 31.62
2 1 2 19.46 2 33.03
2 2 2 17.56 2 33.24
2 3 2 19.74 2 32.08
3 1 2 15.73 2 32.95
3 2 2 17.21 2 33.64
3 3 2 18.09 2 33.40
# Repeated measure analysis
res <- REPEATED_DDCt(
data,
numOfFactors = 1,
numberOfrefGenes = 1,
repeatedFactor = "time",
calibratorLevel = "1",
block = NULL)
# Anova analysis
ANOVA_DDCt(
data,
mainFactor.column = 1,
numOfFactors = 1,
numberOfrefGenes = 1,
block = NULL)
# Paired t.test (equivalent to repeated measure analysis, but not always the same results, due to different calculation methods!)
TTEST_DDCt(
data[1:6,],
numberOfrefGenes = 1,
paired = T)
# Anova analysis
data <- read.csv(system.file("extdata", "data_2factorBlock3ref.csv", package = "rtpcr"))
res <- ANOVA_DDCt(
x = data,
mainFactor.column = 1,
numOfFactors = 2,
numberOfrefGenes = 1,
block = "block",
analyseAllTarget = TRUE)Output
Data output
A lot of outputs including relative expression table, lm models,
residuals, raw data and ANOVA table for each gene can be accessed. The
expression table of all genes is returned by
res$combinedFoldChange. Other outpus for each gene can be
obtained as follow:
| Per_gene Output | Code |
|---|---|
| expression table | res$combinedFoldChange |
| ANOVA table | res$perGene$gene_name$ANOVA_table |
| ANOVA lm | res$perGene$gene_name$lm_ANOVA |
| ANCOVA lm | res$perGene$gene_name$lm_ANCOVA |
| Residuals | resid(res$perGene$gene_name$lm_ANOVA) |
# Relative expression table for the specified column in the input data:
df <- res$combinedFoldChange
df
Relative Expression
gene contrast RE log2FC pvalue sig LCL UCL se Lower.se.RE Upper.se.RE Lower.se.log2FC Upper.se.log2FC
PO R 1.0000 0.0000 1.0000 0.0000 0.0000 0.5506 0.6828 1.4647 0.0000 0.0000
PO S vs R 11.6130 3.5377 0.0001 *** 4.4233 30.4888 0.2286 9.9115 13.6066 3.0193 4.1450
GAPDH R 1.0000 0.0000 1.0000 0.0000 0.0000 0.4815 0.7162 1.3962 0.0000 0.0000
GAPDH S vs R 6.6852 2.7410 0.0001 *** 3.0687 14.5641 0.3820 5.1301 8.7118 2.1034 3.5719
ref2 R 1.0000 0.0000 1.0000 0.0000 0.0000 0.6928 0.6186 1.6164 0.0000 0.0000
ref2 S vs R 0.9372 -0.0936 0.9005 0.3145 2.7929 0.2414 0.7927 1.1079 -0.1107 -0.0792Plot output
A single function of plotFactor is used to produce
barplots for one- to three-factor expression tables.
Plot output: example 1
data <- read.csv(system.file("extdata", "data_3factor.csv", package = "rtpcr"))
#Perform analysis first
res <- ANOVA_DCt(
data,
numOfFactors = 3,
numberOfrefGenes = 1,
block = NULL)
df <- res$combinedResults
df
# Generate three-factor bar plot
p <- plotFactor(
df,
x_col = "SA",
y_col = "log2FC",
group_col = "Type",
facet_col = "Conc",
Lower.se_col = "Lower.se.log2FC",
Upper.se_col = "Upper.se.log2FC",
letters_col = "sig",
letters_d = 0.3,
col_width = 0.7,
dodge_width = 0.7,
fill_colors = c("palegreen3", "skyblue"),
color = "black",
base_size = 14,
alpha = 1,
legend_position = c(0.1, 0.2))
library(ggplot2)
p + theme(
panel.border = element_rect(color = "black", linewidth = 0.5))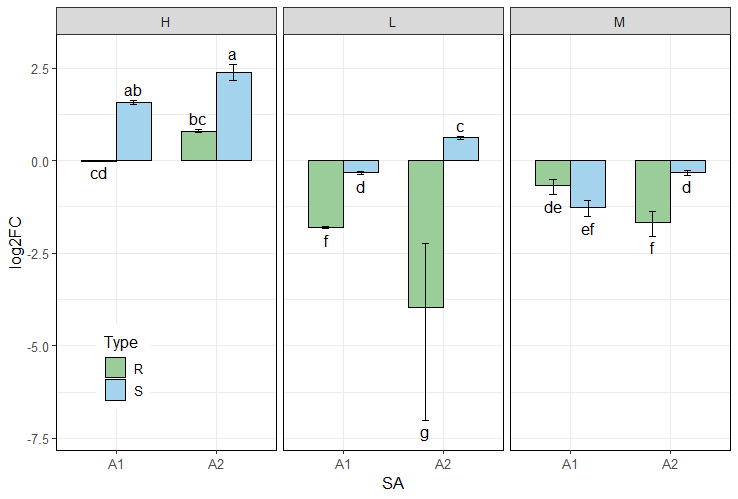
How to edit ouptput plots?
the rtpcr plotFactor function create ggplot objects for
one to three factor table that can furtherbe edited by adding new
layers:
| Task | Example Code |
|---|---|
| Change y-axis label | p + ylab("Relative expression ($\Delta\Delta Ct$ method)") |
| Add a horizontal reference line | p + geom_hline(yintercept = 0, linetype = "dashed") |
| Change y-axis limits | p + scale_y_continuous(expand = expansion(mult = c(0, 0.1))) |
| Relabel x-axis | p + scale_x_discrete(labels = c("A" = "Control", "B" = "Treatment")) |
| Change fill colors | p + scale_fill_brewer(palette = "Set2") |
Plot output: example 2
data <- read.csv(system.file("extdata", "data_2factorBlock.csv", package = "rtpcr"))
res <- ANOVA_DCt(data,
numOfFactors = 2,
block = "block",
numberOfrefGenes = 1)
df <- res$combinedResults
p1 <- plotFactor(
data = df,
x_col = "factor2",
y_col = "RE",
group_col = "factor1",
Lower.se_col = "Lower.se.RE",
Upper.se_col = "Upper.se.RE",
letters_col = "sig",
letters_d = 0.2,
fill_colors = c("aquamarine4", "gold2"),
color = "black",
alpha = 1,
col_width = 0.7,
dodge_width = 0.7,
base_size = 16,
legend_position = c(0.2, 0.8))
library(ggplot2)
p1 +
theme(axis.text.x = element_text(size = 14, color = "black", angle = 45),
axis.text.y = element_text(size = 14,color = "black", angle = 0, hjust = 0.5)) +
theme(legend.text = element_text(colour = "black", size = 14),
legend.background = element_rect(fill = "transparent")) +
scale_y_continuous(expand = expansion(mult = c(0, 0.1)))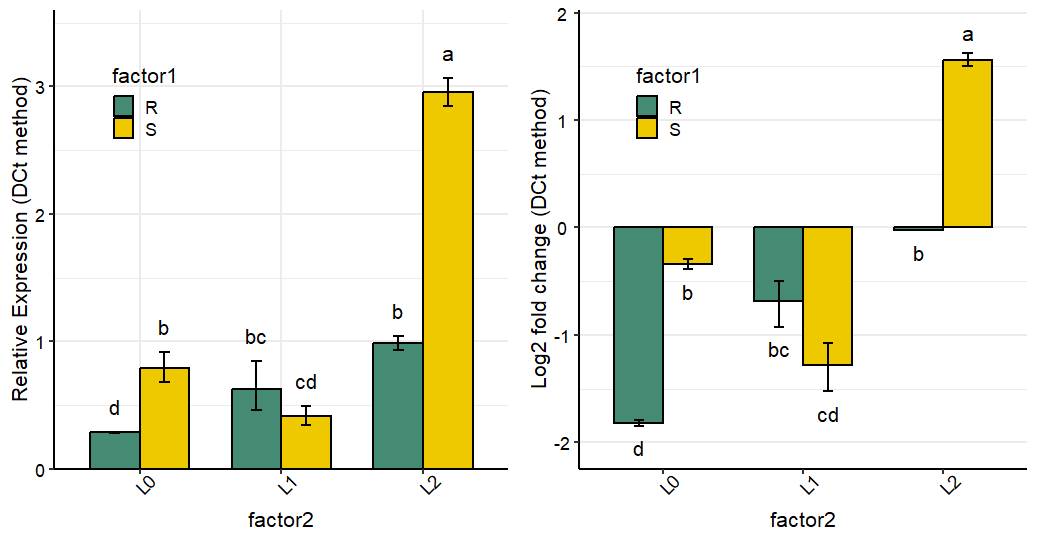
Plot output: example 3
# Heffer et al., 2020, PlosOne
library(dplyr)
df <- read.csv(system.file("extdata", "Heffer2020PlosOne.csv", package = "rtpcr"))
res <- ANOVA_DDCt(
df,
numOfFactors = 1,
mainFactor.column = 1,
numberOfrefGenes = 1,
block = NULL)
data <- res$combinedFoldChange
data$gene <- factor(data$gene, levels = unique(data$gene))
# Selecting only the first words in 'contrast' column to be used as the x-axis labels.
data$contrast <- sub(" .*", "", data$contrast)
# Converting the 'contrast' column as factor and fix the current level order
data$contrast <- factor(data$contrast, levels = unique(data$contrast))
p <- plotFactor(
data = data,
x_col = "contrast",
y_col = "RE",
group_col = "contrast",
facet_col = "gene",
Lower.se_col = "Lower.se.RE",
Upper.se_col = "Upper.se.RE",
letters_col = "sig",
letters_d = 0.2,
alpha = 1,
fill_colors = palette.colors(4, recycle = TRUE),
color = "black",
col_width = 0.5,
dodge_width = 0.5,
base_size = 16,
legend_position = "none")
library(ggplot2)
p + theme(
panel.border = element_rect(color = "black", linewidth = 0.5)) +
theme(axis.text.x = element_text(size = 14, color = "black", angle = 45, hjust = 1)) +
xlab(NULL) +
scale_y_continuous(expand = expansion(mult = c(0, 0.1))) +
theme(
strip.background = element_blank(), # removes the faceting gray background
strip.text = element_text(face = "bold")) # optional: keeps the text visible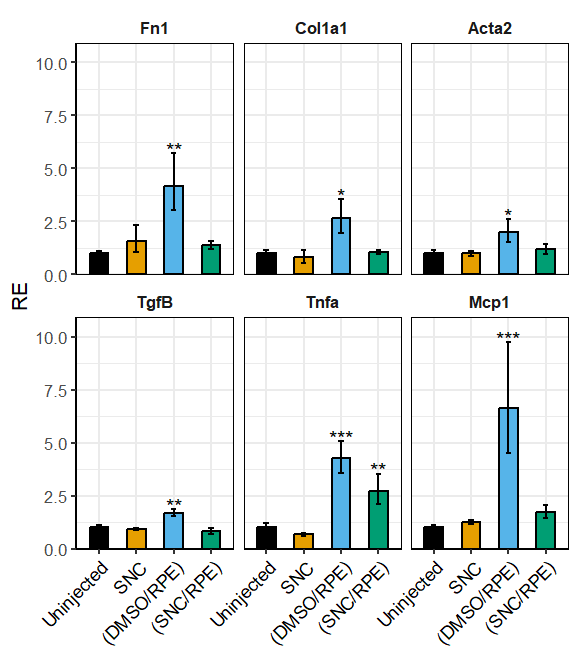
Post-hoc analysis
The Means_DDCt function performs post-hoc comparisons
using a fitted model object produced by ANOVA_DCt,
ANOVA_DDCt or REPEATED_DDCt. It applies
pairwise statistical comparisons of relative expression (RE) values for
user-specified effects via the specs argument. Supported
effects include simple effects, interactions, and slicing, provided the
underlying model is an ANOVA. For ANCOVA models returned by this
package, the Means_DDCt output is limited to simple effects
only.
res <- ANOVA_DDCt(
data_3factor,
numOfFactors = 3,
numberOfrefGenes = 1,
mainFactor.column = 1,
block = NULL)
# Relative expression values for Concentration main effect
Means_DDCt(res$perGene$E_PO$lm_ANOVA, specs = "Conc")
contrast RE SE df LCL UCL p.value sig
L vs H 0.1703610 0.2208988 24 0.1242014 0.2336757 <0.0001 ***
M vs H 0.2227247 0.2208988 24 0.1623772 0.3055004 <0.0001 ***
M vs L 1.3073692 0.2208988 24 0.9531359 1.7932535 0.0928 .
Results are averaged over the levels of: Type, SA
Confidence level used: 0.95
# Relative expression values for Concentration sliced by Type
Means_DDCt(res$perGene$E_PO$lm_ANOVA, specs = "Conc | Type")
Type = R:
contrast RE SE df LCL UCL p.value sig
L vs H 0.103187 0.3123981 24 0.0659984 0.161331 <0.0001 ***
M vs H 0.339151 0.3123981 24 0.2169210 0.530255 <0.0001 ***
M vs L 3.286761 0.3123981 24 2.1022126 5.138776 <0.0001 ***
Type = S:
contrast RE SE df LCL UCL p.value sig
L vs H 0.281265 0.3123981 24 0.1798969 0.439751 <0.0001 ***
M vs H 0.146266 0.3123981 24 0.0935518 0.228684 <0.0001 ***
M vs L 0.520030 0.3123981 24 0.3326112 0.813055 0.0059 **
Results are averaged over the levels of: SA
Confidence level used: 0.95
# Relative expression values for Concentration sliced by Type and SA
Means_DDCt(res$perGene$E_PO$lm_ANOVA, specs = "Conc | Type * SA")Checking normality of residuals
If the residuals from a t.test or an lm or
and lmer object are not normally distributed, the
significance results might be violated. In such cases, non-parametric
tests can be used. For example, the Mann–Whitney test (also known as the
Wilcoxon rank-sum test), implemented via wilcox.test(), is
an alternative to t.test, and kruskal.test() is an
alternative to one-way analysis of variance. These tests assess
differences between population medians using independent samples.
However, the t.test function (along with the
TTEST_DDCt function described above) includes the
var.equal argument. When set to FALSE, perform
t.test under the unequal variances hypothesis. Residuals
for lm (from ANOVA_DCt and
ANOVA_DDCt functions) and lmer (from
REPEATED_DDCt function) objects can be extracted and
plotted as follow:
data <- read.csv(system.file("extdata", "data_repeated_measure_1.csv", package = "rtpcr"))
res3 <- REPEATED_DDCt(
data,
numOfFactors = 1,
numberOfrefGenes = 1,
repeatedFactor = "time",
calibratorLevel = "1",
block = NULL
)
residuals <- resid(res3$perGene$Target$lm)
shapiro.test(residuals)
par(mfrow = c(1,2))
plot(residuals)
qqnorm(residuals)
qqline(residuals, col = "red")Mean of technical replicates
Calculating the mean of technical replicates and generating an output
table suitable for subsequent ANOVA analysis can be accomplished using
the meanTech function. The input dataset must follow the
column structure illustrated in the example data below. Columns used for
grouping should be explicitly specified via the groups
argument of the meanTech function.
# See example input data frame:
data <- read.csv(system.file("extdata", "data_withTechRep.csv", package = "rtpcr"))
data
# Calculating mean of technical replicates
meanTech(data, groups = 1:4)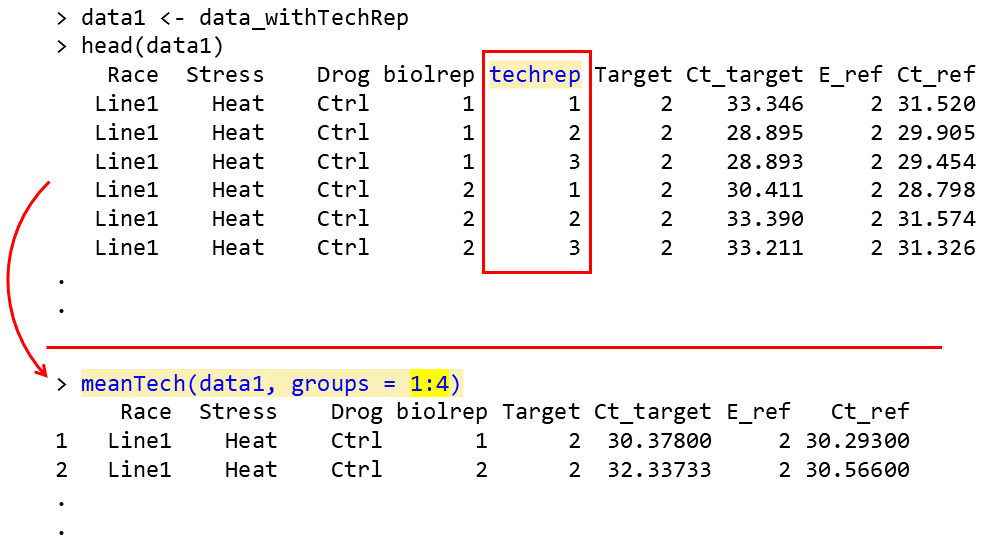
Citation
citation("rtpcr")
To cite the package ‘rtpcr’ in publications, please use:
Ghader Mirzaghaderi (2025). rtpcr: a package for statistical analysis and graphical
presentation of qPCR data in R. PeerJ 13:e20185. https://doi.org/10.7717/peerj.20185
A BibTeX entry for LaTeX users is
@Article{,
title = {rtpcr: A package for statistical analysis and graphical presentation of qPCR data in R},
author = {Ghader Mirzaghaderi},
journal = {PeerJ},
volume = {13},
pages = {e20185},
year = {2025},
doi = {10.7717/peerj.20185},
}Getting help
- If you encounter a clear bug, please file a minimal reproducible example on github
References
Livak, Kenneth J, and Thomas D Schmittgen. 2001. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the Double Delta CT Method. Methods 25 (4). doi.org/10.1006/meth.2001.1262.
Ganger, MT, Dietz GD, Ewing SJ. 2017. A common base method for analysis of qPCR data and the application of simple blocking in qPCR experiments. BMC bioinformatics 18, 1-11. doi.org/10.1186/s12859-017-1949-5.
Mirzaghaderi G. 2025. rtpcr: a package for statistical analysis and graphical presentation of qPCR data in R. PeerJ 13, e20185. doi.org/10.7717/peerj.20185.
Pfaffl MW, Horgan GW, Dempfle L. 2002. Relative expression software tool (REST©) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic acids research 30, e36-e36. doi.org/10.1093/nar/30.9.e36.
Taylor SC, Nadeau K, Abbasi M, Lachance C, Nguyen M, Fenrich, J. 2019. The ultimate qPCR experiment: producing publication quality, reproducible data the first time. Trends in Biotechnology, 37(7), 761-774. doi.org/10.1016/j.tibtech.2018.12.002.
Yuan, JS, Ann Reed, Feng Chen, and Neal Stewart. 2006. Statistical Analysis of Real-Time PCR Data. BMC Bioinformatics 7 (85). doi.org/10.1186/1471-2105-7-85.